- 0
-
 平台客服:赵家玲
平台客服:赵家玲 联系电话:027-81293128
联系电话:027-81293128 -
您的随身业务助手
微信扫一扫 关注「前衍化学」公众号


 发询盘 看报价收询盘 拿订单
发询盘 看报价收询盘 拿订单
微信扫一扫 关注「前衍化学」公众号
发询盘 看报价收询盘 拿订单
图片来源:Veer图库
随着新型冠状病毒肺炎疫情的发展,很多人都在期待,针对此次病毒的特效药能够尽早问世。而最近一系列药物相关进展,也似乎让我们看到了曙光:瑞德西韦已经在武汉进行临床试验,其余几种药物也在体外细胞实验中展现出潜力。但是,对抗新冠病毒的特效药何时才能真正问世?这篇转载自科学大院的文章,讲述了药物研发、生产应该遵循怎样的基本规律与时间要求。
药物研发三步曲
以传统的小分子化学药物为例,新药研发从无到有,要历经药物发现、临床前研究和临床试验“三部曲”,最后才能进入医药市场用于治疗疾病。细说起来可谓步步荆棘,成功者凤毛麟角。
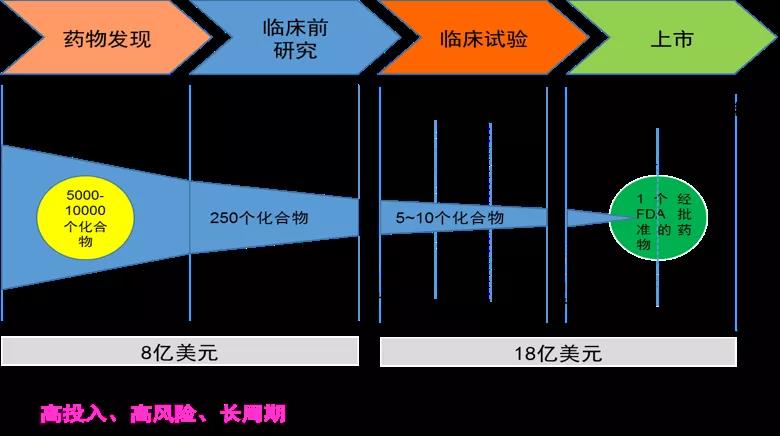
新药研发流程图(图片来源:作者提供)
第一步:候选新药的发现
候选药物的发现首先需要选择和确定药物的作用靶标。靶标是一种与某种疾病发生发展密切相关的生物分子,如蛋白和核酸等,对这种生物分子进行干预,能够治愈或缓解与其相关的疾病。
对于这次的新型冠状病毒而言,准确的药物作用靶标在哪里?
据1月25日的新闻报道,由蒋华良院士和饶子和院士领衔的应急攻关团队快速表达了2019-nCoV水解酶(Mpro)并获得了它的高分辨率晶体结构,从而在此基础上进行药物的筛选。据1月28日的新闻报道,华中科技大学李华教授、沈阳药科大学陈丽霞教授、军事医学研究院李行舟研究员等组成联合攻关小组,发现了冠状病毒nsp3编码的木瓜样蛋白酶(papain-like protease,PLP)在病毒基因组复制及逃避宿主抗病毒天然免疫中发挥重要作用,是药物开发的良好靶点。更多科学家目前正在寻找更多可能抑制2019-nCoV的药物靶点,为抗新型冠状病毒肺炎药物的研发提供更多指导信息。
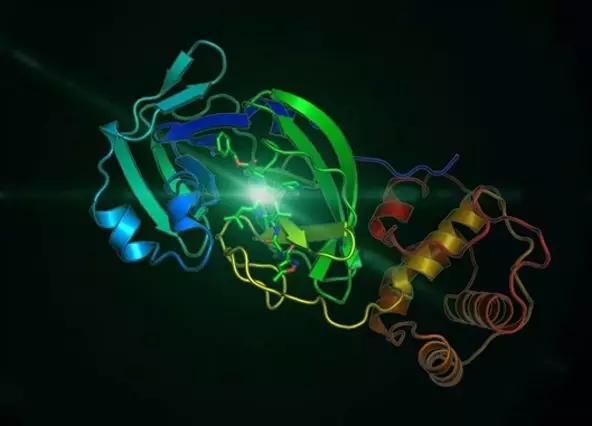
2019-nCoV Mpro晶体结构 (图片来源:中国科学院网站)
药物作用的靶标确定之后,药物化学家们需要根据靶标的空间结构,设计或者合成有作用的先导化合物。这个过程很关键,也考验药物化学家的能力,这些化合物可以是全新结构的化合物,也可以来自天然产物(动物、植物、海洋生物),甚至还可以是一些已经上市的药物。
经活性筛选得到先导化合物后,还需要以先导化合物为模板合成大量的新化合物,以进行构效关系研究,进一步筛选优化得到活性更好的化合物,同时还得系统地研究化合物的理化性质,代谢性质以及毒理早期数据,才能筛选出来满足成药性的最优化合物,这时候可以作为候选药物,进入临床前开发。
第二步:候选新药临床前研究
确定候选药物是新药研发的基石,接下来新药就从研究进入了开发阶段,也就是系统的临床前和临床研究工作,这时候需要大量的资金投入这个“主角”身上。
临床前研究需要进行包括原料药和制剂的药学研究,动物体内的药理药效,药代动力学,以及安全性评价在内的系统研究工作,这部分研究需要在动物身上进行。
药学研究:分为原料药和制剂两部分工作。早期合成的原料药主要用于药效、药代、早期毒理等的研究。这期间需要对原料药的杂质(包括基因毒性杂质)进行系统的研究,从而充分地评价药物及其相关杂质的安全性。
随着新药研发进入后面的临床阶段,药物化学家们还得不断地放大合成的规模,优化开发更加合理的生产工艺,并且符合GMP(Good Manufacturing Practice)生产的要求,逐步满足将来商业化生产的需求。
制剂部门需要进行系统的处方和工艺研究,质量标准和控制研究,稳定性研究等工作,从而开发出符合临床需求的新药制剂。
药效学研究:这阶段主要是为了解答药物作用的机制,包括了解药物多大剂量起效,以及在什么时候有效,对什么人群有效等相关科学问题,从而确定药物的给药剂量和频率等关键信息。
而解决这些问题的前提,是要找到合适的体内体外评价模型,也就是找到合适的分子、细胞进行体外药效评价。(编者注:最近,阿比朵尔等药物的体外细胞实验,即属于该阶段)此后,需要构建对症的动物模型进行体内药效评价。而对症的动物模型往往很难构建,容易成为新药研发的瓶颈。对于这次的新型冠状病毒肺炎,加快新药研发的步伐离不开对症动物模型的尽早建立。
药代动力学研究:研究药物在动物体内的吸收、分布、代谢、排泄的性质(ADME),从而指导临床研究以何种形式给药。一般优先选择口服,如果口服不吸收,则考虑注射或者吸入给药。另一方面,药代动力学也有助于确定新药的给药频率和剂量,例如一天需要给药几次。
安全性评价:一般需要在至少两种动物种属(例如大鼠和犬)中进行评价,包括急毒,亚急毒,长期毒性,安全药理,遗传毒性,生殖毒性,致癌性,致敏性,依赖性等。从而获得毒性靶器官,最大耐受剂量,未见明显毒性反应剂量等关键结果。这些毒性评价的结果可为确定新药的首次人体试验的起始剂量提供依据,并为制定人体临床试验的风险防控措施提供依据。对于一些需要长期服用治疗慢性病的新药,则需要进行更长时间的毒理实验研究。
第三步:临床研究
在完成了系统的临床前研究后,接下来就是进入临床阶段了,临床阶段需要在人体上进行试验,因此药物进入临床研究前必须得到国家药品监督管理部门的审批。在中国,新药的研发机构需要向国家药品监督管理局(NMPA)提交新药临床申请,获得许可后才能进行人体临床试验。
临床研究还需要分为四个阶段:Ⅰ期临床试验,Ⅱ期临床试验,Ⅲ期临床试验和IV期临床研究(药物上市后监测)。
Ⅰ期临床试验
在健康志愿者(对于肿瘤药物而言通常为肿瘤患者)身上研究新药在人体内的安全耐受性和药物代谢性质的试验,为制定给药方案和推荐安全剂量提供依据。
Ⅱ期临床试验
在真正的患者身上进行临床试验,主要目的是获得药物治疗的有效性数据,以及进一步的安全性数据,一般这个时候可以明确具体的适应症。
Ⅲ期临床试验
在更大范围的病人志愿者身上进行扩大的多中心临床试验,是治疗作用的确证阶段,也是决定药物研发是否能够成功的关键阶段。
提交新药上市申请
在完成了上面三个阶段的临床试验后,对临床数据进行统计学分析,证明药物安全有效的同时,确保新药质量可控,也就是安全,有效,可控这三点都符合要求后,药品上市许可持有人(也就是药企或者研发机构),向药监部门提交新药上市申请。获得药监部门批准后,新药才可上市销售,供医生和病人选择。
Ⅳ期临床试验(药物上市后监测)
监测药物的长期副作用等情况。如果新药上市后发现了新的严重不良反应,比如心脏毒性等等,该药有可能会被药监部门撤市。
特殊时刻,仍要遵循新药研发规律
看到这里,很多人可能会有疑惑,紧急关头,我们能否缩短药物研发的时间、降低标准?
答案是,可以加快研发速度,但仍要遵循新药研发规律。药物的研发是一项周期长,投资高,风险大的系统工程,每一个环节都来不得半点错误,否则人命关天。
周期长:一个创新药物从实验室研究到最终上市可能需要10年;
投资高:据不完全统计,全球的各大制药公司对于一个创新药物的资金投入,从最初到研发上市花费金额平均高达20多亿美金;
风险大:导致药物研发失败的原因有很多,如在人体内的有效性不够、人体无法耐受或有严重毒副作用、药物在人体内的吸收/分布/代谢/排泄的不良性质等。从实验室研发出潜在有效的化合物到最终在临床确定有效,并能够应用到市场的药物,可能1万个活性化合物苗子中才能有一个化合物最终能成功上市。
那么,有些已经上市的药物,或者已经在国外做临床试验的新药,是不是可以弯道超车,加快研发进程呢?
对于已经上市或者国内外正处于临床试验阶段的药物,如果需要增加新的适应症,例如将最初为抗埃博拉病毒设计的药物(瑞德西韦)用于治疗新冠肺炎,这种情况也是需要经过系统的临床试验研究才能确定能否用于该新适应症的,而且都需要与国家药品监督管理局药品审评中心进行沟通交流,并且得到药监局的许可才能进行相关的临床试验。(编者注:针对瑞德西韦的III期临床试验已经于2月6日在武汉开展。据介绍,临床试验最快可能在4月底得出结果。)
由于新型冠状病毒和SARS有类似之处,同时还有同为冠状病毒的中东呼吸综合征(MERS)的治疗经验,相比17年前的SARS,科学家们对新型冠状病毒肺炎的研究和理解要深入得多,某些程度上可能会缩减药物研发的时间,但是,新药开发的规律无法被逾越,药物研发这事,急不来。
关键词:特效药
分享至:
![]()
![]()




鄂公网安备 42011102004299号
© 2014-2024 前衍化学科技(武汉)有限公司 版权所有 鄂ICP备20009754号-1

